Doxorubicin Other names: Doxorubicin hydrochloride
Chemical formula: C₂₇H₂₉NO₁₁ Molecular mass: 543.519 g/mol PubChem compound: 31703
Interactions
Doxorubicin interacts in the following cases:
Hepatic impairment
Doxorubicin pharmacokinetics determined in a small number of patients with elevated total bilirubin levels do not differ from patients with normal total bilirubin; however, until further experience is gained, the doxorubicin dosage in patients with impaired hepatic function should be reduced based on the experience from the breast and ovarian clinical trial programs as follows: at initiation of therapy, if the bilirubin is between 1.2-3.0 mg/dl, the first dose is reduced by 25%. If the bilirubin is >3.0 mg/dl, the first dose is reduced by 50%. If the patient tolerates the first dose without an increase in serum bilirubin or liver enzymes, the dose for cycle 2 can be increased to the next dose level, i.e., if reduced by 25% for the first dose, increase to full dose for cycle 2; if reduced by 50% for the first dose, increase to 75% of full dose for cycle 2. The dosage can be increased to full dose for subsequent cycles if tolerated. Doxorubicin can be administered to patients with liver metastases with concurrent elevation of bilirubin and liver enzymes up to 4 x the upper limit of the normal range. Prior to doxorubicin administration, evaluate hepatic function using conventional clinical laboratory tests such as ALT/AST, alkaline phosphatase, and bilirubin.
Cyclophosphamide
In patients with AIDS, exacerbation of cyclophosphamide-induced haemorrhagic cystitis have been reported with standard doxorubicin hydrochloride.
Mercaptopurine
In patients with AIDS, enhancement of the hepatotoxicity of 6-mercaptopurine have been reported with standard doxorubicin hydrochloride.
Myelosuppression
Many patients treated with doxorubicin have baseline myelosuppression due to such factors as their pre-existing HIV disease or numerous concomitant or previous medications, or tumours involving bone marrow. In the pivotal trial in patients with ovarian cancer treated at a dose of 50 mg/m², myelosuppression was generally mild to moderate, reversible, and was not associated with episodes of neutropaenic infection or sepsis. Moreover, in a controlled clinical trial of doxorubicin vs. topotecan, the incidence of treatment related sepsis was substantially less in the doxorubicin-treated ovarian cancer patients as compared to the topotecan treatment group. A similar low incidence of myelosuppression was seen in patients with metastatic breast cancer receiving doxorubicin in a first-line clinical trial. In contrast to the experience in patients with breast cancer or ovarian cancer, myelosuppression appears to be the dose-limiting adverse event in patients with AIDS-KS. Because of the potential for bone marrow suppression, periodic blood counts must be performed frequently during the course of doxorubicin therapy, and at a minimum, prior to each dose of doxorubicin.
Persistent severe myelosuppression, may result in superinfection or haemorrhage.
In controlled clinical studies in patients with AIDS-KS against a bleomycin/vincristine regimen, opportunistic infections were apparently more frequent during treatment with doxorubicin. Patients and doctors must be aware of this higher incidence and take action as appropriate.
Palmar-Plantar erythrodysesthesia (PPE)
To manage adverse events such as palmar-plantar erythrodysesthesia (PPE), the dose may be reduced or delayed. Guidelines for doxorubicin dose modification secondary to these adverse effects are provided in the table below. The toxicity grading in these tables is based on the National Cancer Institute Common Toxicity Criteria (NCI-CTC).
Palmar-Plantar erythrodysesthesia:
| Week after prior doxorubicin dose | |||
|---|---|---|---|
| Toxicity grade at current assessment | Week 4 | Week 5 | Week 6 |
| Grade 1 (mild erythema, swelling, or desquamation not interfering with daily activities) | Redose unless patient has experienced a previous grade 3 or 4 skin toxicity, in which case wait an additional week | Redose unless patient has experienced a previous grade 3 or 4 skin toxicity, in which case wait an additional week | Decrease dose by 25%; return to 4 week interval |
| Grade 2 (erythema, desquamation, or swelling interfering with, but not precluding normal physical activities; small blisters or ulcerations less than 2 cm in diameter) | Wait an additional week | Wait an additional week | Decrease dose by 25%; return to 4 week interval |
| Grade 3 (blistering, ulceration, or swelling interfering with walking or normal daily activities; cannot wear regular clothing) | Wait an additional week | Wait an additional week | Wait an additional week |
| Grade 4 (diffuse or local process causing infectious complications, or a bedridden state or hospitalisation) | Wait an additional week | Wait an additional week | Withdraw patient |
Neutropenia, thrombopenia
To manage adverse events such as haematological toxicity, the dose may be reduced or delayed. Guidelines for doxorubicin dose modification secondary to these adverse effects are provided in the table below. The toxicity grading in these tables is based on the National Cancer Institute Common Toxicity Criteria (NCI-CTC).
The table for haematological toxicity provides the schedule followed for dose modification in clinical trials in the treatment of patients with breast or ovarian cancer only.
Haematological toxicity (ANC or platelets) - Management of patients with breast or ovarian cancer:
| GRADE | ANC | PLATELETS | MODIFICATION |
|---|---|---|---|
| Grade 1 | 1,500 - 1,900 | 75,000 - 150,000 | Resume treatment with no dose reduction. |
| Grade 2 | 1,000 - <1.500 | 50,000 - <75,000 | Wait until ANC ≥1,500 and platelets ≥75,000, redose with no dose reduction. |
| Grade 3 | 500 - <1,000 | 25,000 - <50,000 | Wait until ANC ≥1,500 and platelets ≥75,000, redose with no dose reduction. |
| Grade 4 | <500 | <25,000 | Wait until ANC ≥1.500 and platelets ≥75,000; decrease dose by 25% or continue full dose with growth factor support. |
Stomatitis
To manage adverse events such as stomatitis, the dose may be reduced or delayed. Guidelines for doxorubicin dose modification secondary to these adverse effects are provided in the table below. The toxicity grading in these tables is based on the National Cancer Institute Common Toxicity Criteria (NCI-CTC).
Stomatitis:
| Week after prior doxorubicin dose | |||
|---|---|---|---|
| Toxicity grade at current assessment | Week 4 | Week 5 | Week 6 |
| Grade 1 (painless ulcers, erythema, or mild soreness) | Redose unless patient has experienced a previous grade 3 or 4 stomatitis in which case wait an additional week | Redose unless patient has experienced a previous grade 3 or 4 stomatitis in which case wait an additional week | Decrease dose by 25%; return to 4 week interval or withdraw patient per physician's assessment |
| Grade 2 (painful erythema, oedema, or ulcers, but can eat) | Wait an additional week | Wait an additional week | Decrease dose by 25%; return to 4 week interval or withdraw patient per physician's assessment |
| Grade 3 (painful erythema, edema, or ulcers, but cannot eat) | Wait an additional week | Wait an additional week | Withdraw patient |
| Grade 4 (requires parenteral or enteral support) | Wait an additional week | Wait an additional week | Withdraw patient |
Cardiac toxicity
It is recommended that all patients receiving doxorubicin routinely undergo frequent ECG monitoring. Transient ECG changes such as T-wave flattening, S-T segment depression and benign arrhythmias are not considered mandatory indications for the suspension of doxorubicin therapy. However, reduction of the QRS complex is considered more indicative of cardiac toxicity. If this change occurs, the most definitive test for anthracycline myocardial injury, i.e. endomyocardial biopsy, must be considered.
More specific methods for the evaluation and monitoring of cardiac functions as compared to ECG are a measurement of left ventricular ejection fraction by echocardiography or preferably by Multigated
Angiography (MUGA). These methods must be applied routinely before the initiation of doxorubicin therapy and repeated periodically during treatment. The evaluation of left ventricular function is considered to be mandatory before each additional administration of doxorubicin that exceeds a lifetime cumulative anthracycline dose of 450 mg/m².
The evaluation tests and methods mentioned above concerning the monitoring of cardiac performance during anthracycline therapy are to be employed in the following order: ECG monitoring, measurement of left ventricular ejection fraction, endomyocardial biopsy. If a test result indicates possible cardiac injury associated with doxorubicin therapy, the benefit of continued therapy must be carefully weighed against the risk of myocardial injury.
In patients with cardiac disease requiring treatment, administer doxorubicin only when the benefit outweighs the risk to the patient.
Exercise caution in patients with impaired cardiac function who receive doxorubicin.
Whenever cardiomyopathy is suspected, i.e. the left ventricular ejection fraction has substantially decreased relative to pre-treatment values and/or left ventricular ejection fraction is lower than a prognostically relevant value (e.g., <45%), endomyocardial biopsy may be considered and the benefit of continued therapy must be carefully evaluated against the risk of developing irreversible cardiac damage.
Congestive heart failure due to cardiomyopathy may occur suddenly, without prior ECG changes and may also be encountered several weeks after discontinuation of therapy.
Caution must be observed in patients who have received other anthracyclines. The total dose of doxorubicin hydrochloride must also take into account any previous (or concomitant) therapy with cardiotoxic compounds such as other anthracyclines/anthraquinones or e.g. 5-fluorouracil. Cardiac toxicity also may occur at cumulative anthracycline doses lower than 450 mg/m² in patients with prior mediastinal irradiation or in those receiving concurrent cyclophosphamide therapy.
The cardiac safety profile for the dosing schedule recommended for both breast and ovarian cancer (50 mg/m²) is similar to the 20 mg/m² profile in patients with AIDS-KS.
Pregnancy
Doxorubicin is suspected to cause serious birth defects when administered during pregnancy. Therefore, doxorubicin should not be used during pregnancy unless clearly necessary.
Nursing mothers
It is not known whether doxorubicin is excreted in human milk. Because many medicinal products, including anthracyclines, are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants, therefore mothers must discontinue nursing prior to beginning doxorubicin treatment. Health experts recommend that HIV infected women do not breast-feed their infants under any circumstances in order to avoid transmission of HIV.
Carcinogenesis, mutagenesis and fertility
Women of child-bearing potential
Women of child-bearing potential must be advised to avoid pregnancy while they or their male partner are receiving doxorubicin and in the six months following discontinuation of doxorubicin therapy.
Fertility
The effect of doxorubicin on human fertility has not been evaluated.
Effects on ability to drive and use machines
Doxorubicin has no or negligible influence on the ability to drive and use machines. However, in clinical studies to date, dizziness and somnolence were associated infrequently (<5%) with the administration of doxorubicin. Patients who suffer from these effects must avoid driving and operating machinery.
Adverse reactions
Summary of the safety profile
The most common undesirable effect reported in breast/ovarian clinical trials (50 mg/m² every 4 weeks) was palmar-plantar erythrodysesthesia (PPE). The overall incidence of PPE reported was 44.0%-46.1%. These effects were mostly mild, with severe (grade 3) cases reported in 17%-19.5%. The reported incidence of life-threatening (grade 4) cases was <1%. PPE infrequently resulted in permanent treatment discontinuation (3.7%-7.0%). PPE is characterised by painful, macular reddening skin eruptions. In patients experiencing this event, it is generally seen after two or three cycles of treatment. Improvement usually occurs in one - two weeks, and in some cases, may take up to 4 weeks or longer for complete resolution. Pyridoxine at a dose of 50-150 mg per day and corticosteroids have been used for the prophylaxis and treatment of PPE, however, these therapies have not been evaluated in phase III trials. Other strategies to prevent and treat PPE include keeping hands and feet cool, by exposing them to cool water (soaks, baths, or swimming), avoiding excessive heat/hot water and keeping them unrestricted (no socks, gloves, or shoes that are tight fitting). PPE appears to be primarily related to the dose schedule and can be reduced by extending the dose interval 1-2 weeks. However, this reaction can be severe and debilitating in some patients and may require discontinuation of treatment. Stomatitis/mucositis and nausea were also commonly reported in breast/ovarian cancer patient populations, whereas the AIDS-KS Program (20 mg/m² every 2 weeks), myelosuppression (mostly leukopaenia) was the most common side effect (see AIDS-KS). PPE was reported in 16% of multiple myeloma patients treated with doxorubicin plus bortezomib combination therapy. Grade 3 PPE was reported in 5% of patients. No grade 4 PPE was reported. The most frequently reported (medicine-related treatment-emergent) adverse events in combination therapy (doxorubicin + bortezomib) were nausea (40%), diarrhoea (35%), neutropaenia (33%), thrombocytopaenia (29%), vomiting (28%), fatigue (27%), and constipation (22%).
Breast cancer program
509 patients with advanced breast cancer who had not received prior chemotherapy for metastatic disease were treated with doxorubicin (n=254) at a dose of 50 mg/m² every 4 weeks, or doxorubicin (n=255) at a dose of 60 mg/m² every 3 weeks, in a phase III clinical trial (I97-328). The following common adverse events were reported more often with doxorubicin than with doxorubicin: nausea (53% vs. 37%; grade ¾ 5% vs. 3%), vomiting (31% vs. 19%; grade ¾ 4% vs. less than 1%), any alopecia (66% vs. 20%), pronounced alopecia (54% vs.7%), and neutropaenia (10% vs. 4%; grade ¾ 8% vs. 2%).
Mucositis (23% vs. 13%; grade ¾ 4% vs. 2%), and stomatitis (22% vs. 15%; grade ¾ 5% vs. 2%) were reported more commonly with doxorubicin than with doxorubicin. The average duration of the most common severe (grade ¾) events for both groups was 30 days or less. See Table 1 for complete listing of undesirable effects reported in doxorubicin-treated patients.
The incidence of life threatening (grade 4) haematologic effects was <1.0% and sepsis was reported in 1% of patients. Growth factor support or transfusion support was necessary in 5.1 % and 5.5% of patients, respectively.
Clinically significant laboratory abnormalities (grades 3 and 4) in this group was low with elevated total bilirubin, AST and ALT reported in 2.4%, 1.6% and <1% of patients respectively. No clinically significant increases in serum creatinine were reported.
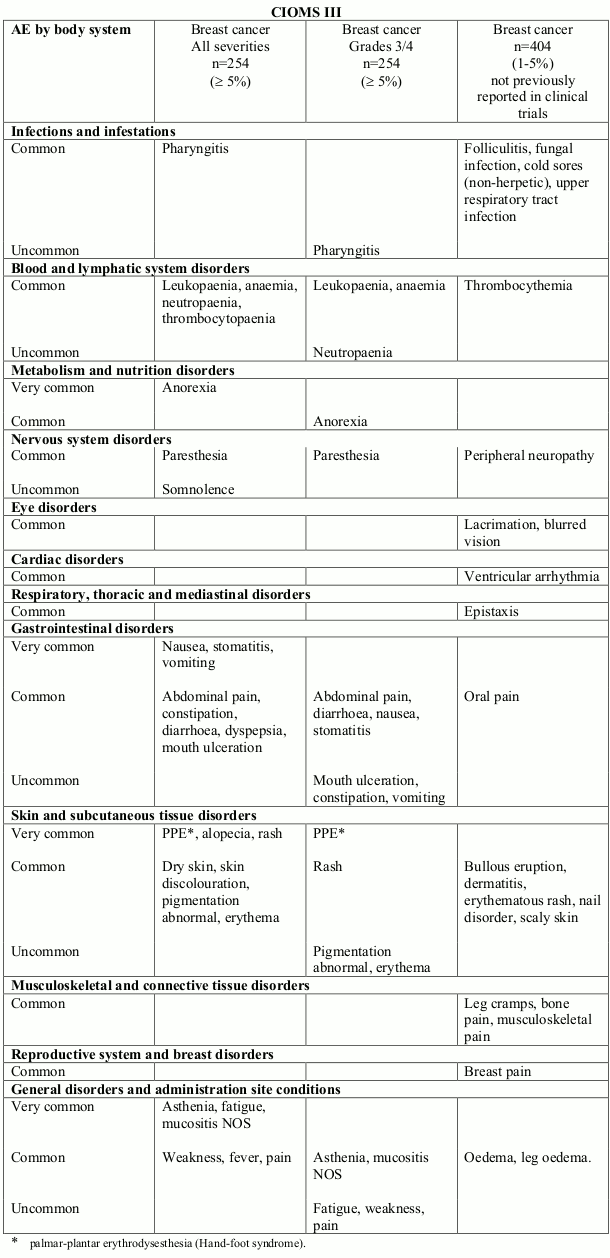
Table 1. Treatment related undesirable effects reported in breast cancer clinical trials (50 mg/m² every 4 weeks) (doxorubicin-treated patients) by severity, MedDRA system organ class and preferred term:
Very common (≥1/10); Common (≥1/100, <1/10); Uncommon (≥1/1,000, <1/100)
Ovarian cancer program
512 patients with ovarian cancer (a subset of 876 solid tumour patients) were treated with doxorubicin at a dose of 50 mg/m² in clinical trials. See Table 2 for undesirable effects reported in doxorubicin-treated patients.
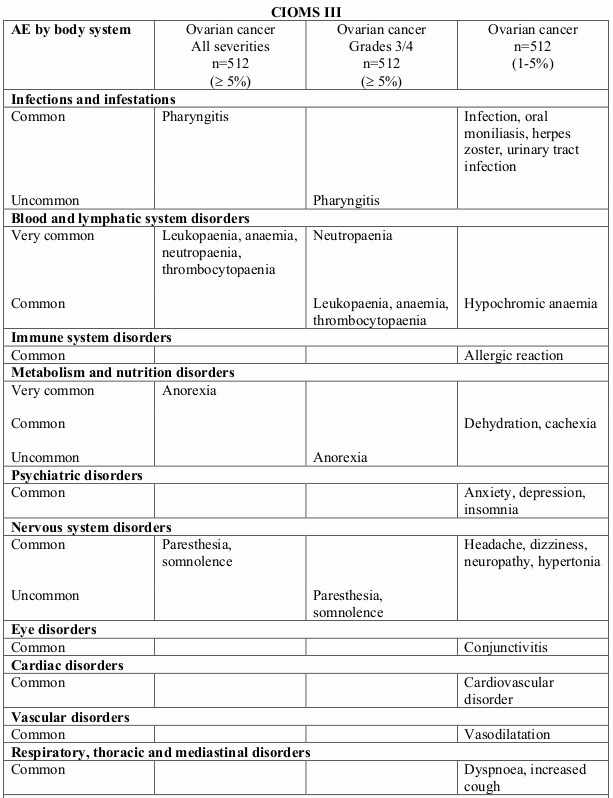
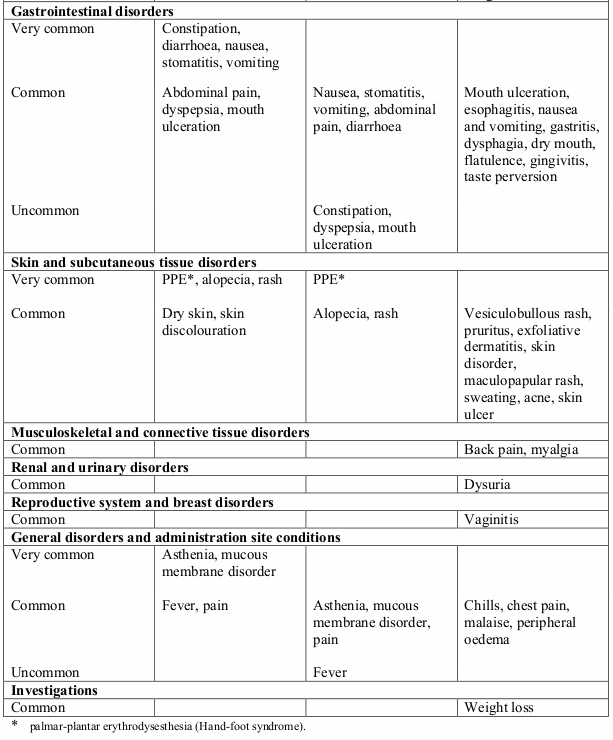
Table 2. Treatment related undesirable effects reported in ovarian cancer clinical trials (50 mg/m² every 4 weeks) (doxorubicin-treated patients) by severity, MedDRA system organ class and preferred term:
Very common (≥1/10); Common (≥1/100, <1/10); Uncommon (≥1/1,000, <1/100)
Myelosuppression was mostly mild or moderate and manageable. Sepsis related to leukopaenia was observed infrequently (<1%). Growth factor support was required infrequently (<5%) and transfusion support was required in approximately 15% of patients.
In a subset of 410 patients with ovarian cancer, clinically significant laboratory abnormalities occurring in clinical trials with doxorubicin included increases in total bilirubin (usually in patients with liver metastases) (5%) and serum creatinine levels (5%). Increases in AST were less frequently (<1%) reported.
Solid tumour patients: in a larger cohort of 929 patients with solid tumours (including breast cancer and ovarian cancer) predominantly treated at a dose of 50 mg/m² every 4 weeks, the safety profile and incidence of adverse effects are comparable to those of the patients treated in the pivotal breast cancer and ovarian cancer trials.
Multiple myeloma program
Of 646 patients with multiple myeloma who have received at least 1 prior therapy, 318 patients were treated with combination therapy of doxorubicin 30 mg/m² as a one hour intravenous infusion administered on day 4 following bortezomib which is administered at 1.3 mg/m² on days 1, 4, 8, and 11, every three
weeks or with bortezomib monotherapy in a phase III clinical trial. See Table 3 for adverse effects reported in >5% patients treated with combination therapy of doxorubicin plus bortezomib.
Neutropaenia, thrombocytopaenia, and anaemia were the most frequently reported haematologic events reported with both combination therapy of doxorubicin plus bortezomib and bortezomib monotherapy. The incidence of grade 3 and 4 neutropaenia was higher in the combination therapy group than in the monotherapy group (28% vs. 14%). The incidence of grade 3 and 4 thrombocytopaenia was higher in the combination therapy group than in the monotherapy group (22% vs. 14%). The incidence of anaemia was similar in both treatment groups (7% vs. 5%).
Stomatitis was reported more frequently in the combination therapy group (16%) than in the monotherapy group (3%), and most cases were grade 2 or less in severity. Grade 3 stomatitis was reported in 2% of patients in the combination therapy group. No grade 4 stomatitis was reported.
Nausea and vomiting were reported more frequently in the combination therapy group (40% and 28%) than in the monotherapy group (32% and 15%) and were mostly grade 1 and 2 in severity.
Treatment discontinuation of one or both agents due to adverse events was seen in 38% of patients. Common adverse events which led to treatment discontinuation of bortezomib and doxorubicin included PPE, neuralgia, peripheral neuropathy, peripheral sensory neuropathy, thrombocytopaenia, decreased ejection fraction, and fatigue.
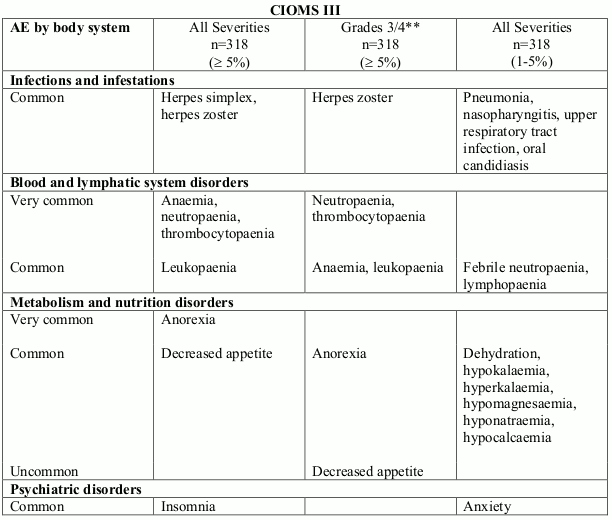
Table 3. Treatment related undesirable effects reported in multiple myeloma clinical trial (doxorubicin 30 mg/m² in combination with bortezomib every 3 weeks) by severity, MedDRA system organ class and preferred term:
Very common (≥1/10); Common (≥1/100, <1/10); Uncommon (≥1/1,000, <1/100)
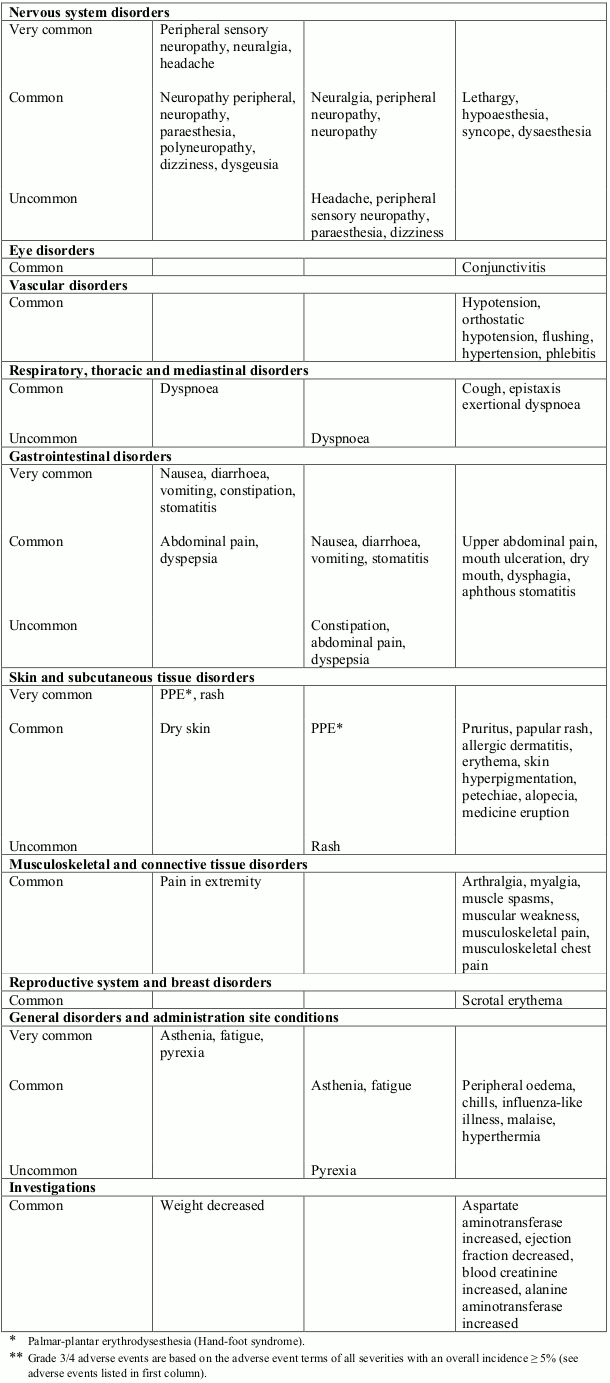
AIDS-related KS program
Clinical studies on AIDS-KS patients treated at 20 mg/m² with doxorubicin show that myelosuppression was the most frequent undesirable effect considered related to doxorubicin occurring very commonly (in approximately one-half of the patients).
Leukopaenia is the most frequent undesirable effect experienced with doxorubicin in this population; neutropaenia, anaemia and thrombocytopaenia have been observed. These effects may occur early on in treatment. Haematological toxicity may require dose reduction or suspension or delay of therapy. Temporarily suspend doxorubicin treatment in patients when the ANC count is <1,000/mm³ and/or the platelet count is <50,000/mm³. G-CSF (or GM-CSF) may be given as concomitant therapy to support the blood count when the ANC count is <1,000/mm³ in subsequent cycles. The haematological toxicity for ovarian cancer patients is less severe than in the AIDS-KS setting (see section for ovarian cancer patients above).
Respiratory undesirable effects commonly occurred in clinical studies of doxorubicin and may be related to opportunistic infections (OI's) in the AIDS population. Opportunistic infections are observed in KS patients after administration with doxorubicin, and are frequently observed in patients with HIV-induced immunodeficiency. The most frequently observed OI's in clinical studies were candidiasis, cytomegalovirus, herpes simplex, Pneumocystis carinii pneumonia, and mycobacterium avium complex.
Table 4. Undesirable effects observed in patients with AIDS-related KS according to CIOMS III frequency categories:
Very common (≥1/10); Common (≥1/100, <1/10); Uncommon (≥1/1,000, <1/100)
Infections and infestations
common: oral moniliasis
Blood and lymphatic system disorders
very common: neutropaenia, anaemia, leukopaenia
common: thrombocytopaenia
Metabolism and nutrition disorders
common: anorexia
Psychiatric disorders
uncommon: confusion
Nervous system disorders
common: dizziness
uncommon: paresthesia
Eye disorders
common: retinitis
Vascular disorders
common: vasodilatation
Respiratory, thoracic and mediastinal disorders
common: dyspnoea
Gastrointestinal disorders
very common: nausea
common: diarrhoea, stomatitis, vomiting, mouth ulceration, abdominal pain, glossitis, constipation, nausea and vomiting
Skin and subcutaneous tissue disorders
common: alopecia, rash
uncommon: palmar-plantar erythrodysesthesia (PPE)
General disorders and administration site conditions
common: asthenia, fever, infusion-associated acute reactions
Investigations
common: weight loss
Other less frequently (<5%) observed undesirable effects included hypersensitivity reactions including anaphylactic reactions. Following marketing, bullous eruption has been reported rarely in this population.
Clinically significant laboratory abnormalities frequently (>5%) occurred including increases in alkaline phosphatase; AST and bilirubin which were believed to be related to the underlying disease and not doxorubicin. Reduction in haemoglobin and platelets were less frequently (<5%) reported. Sepsis related to leukopaenia was rarely (<1%) observed. Some of these abnormalities may have been related to the underlying HIV infection and not doxorubicin.
All patients
100 out of 929 patients (10.8%) with solid tumours were described as having an infusion-associated reaction during treatment with doxorubicin as defined by the following Costart terms: allergic reaction, anaphylactoid reaction, asthma, face oedema, hypotension, vasodilatation, urticaria, back pain, chest pain, chills, fever, hypertension, tachycardia, dyspepsia, nausea, dizziness, dyspnoea, pharyngitis, rash, pruritus, sweating, injection site reaction and medicinal product interaction. Permanent treatment discontinuation was infrequently reported at 2%. A similar incidence of infusion reactions (12.4%) and treatment discontinuation (1.5%) was observed in the breast cancer program. In patients with multiple myeloma receiving doxorubicin plus bortezomib, infusion-associated reactions have been reported at a rate of 3%. In patients with AIDS-KS, infusion-associated reactions, were characterised by flushing, shortness of breath, facial oedema, headache, chills, back pain, tightness in the chest and throat and/or hypotension and can be expected at the rate of 5% to 10%. Very rarely, convulsions have been observed in relation to infusion reactions. In all patients, infusion-associated reactions occurred primarily during the first infusion. Temporarily stopping the infusion usually resolves these symptoms without further therapy. In nearly all patients, doxorubicin treatment can be resumed after all symptoms have resolved without recurrence. Infusion reactions rarely recur after the first treatment cycle with doxorubicin.
Myelosuppression associated with anaemia, thrombocytopaenia, leukopaenia, and rarely febrile neutropaenia, has been reported in doxorubicin-treated patients.
Stomatitis has been reported in patients receiving continuous infusions of conventional doxorubicin hydrochloride and was frequently reported in patients receiving doxorubicin. It did not interfere with patients completing therapy and no dosage adjustments are generally required, unless stomatitis is affecting a patient's ability to eat. In this case, the dose interval may be extended by 1-2 weeks or the dose reduced.
An increased incidence of congestive heart failure is associated with doxorubicin therapy at cumulative lifetime doses >450 mg/m² or at lower doses for patients with cardiac risk factors. Endomyocardial biopsies on nine of ten AIDS-KS patients receiving cumulative doses of doxorubicin greater than 460 mg/m² indicate no evidence of anthracycline-induced cardiomyopathy. The recommended dose of doxorubicin for AIDS-KS patients is 20 mg/m² every two-to-three weeks. The cumulative dose at which cardiotoxicity would become a concern for these AIDS-KS patients (>400 mg/m²) would require more than 20 courses of doxorubicin therapy over 40 to 60 weeks.
In addition, endomyocardial biopsies were performed in 8 solid tumour patients with cumulative anthracycline doses of 509 mg/m²-1,680 mg/m². The range of Billingham cardiotoxicity scores was grades 0-1.5. These grading scores are consistent with no or mild cardiac toxicity.
In the pivotal phase III trial versus doxorubicin, 58/509 (11.4%) randomised subjects (10 treated with doxorubicin at a dose of 50 mg/m²/every 4 weeks versus 48 treated with doxorubicin at a dose of 60 mg/m²/every 3 weeks) met the protocol-defined criteria for cardiac toxicity during treatment and/or follow-up. Cardiac toxicity was defined as a decrease of 20 points or greater from baseline if the resting LVEF remained in the normal range or a decrease of 10 points or greater if the LVEF became abnormal (less than the lower limit for normal). None of the 10 doxorubicin subjects who had cardiac toxicity by LVEF criteria developed signs and symptoms of CHF. In contrast, 10 of 48 doxorubicin subjects who had cardiac toxicity by LVEF criteria also developed signs and symptoms of CHF.
In patients with solid tumours, including a subset of patients with breast and ovarian cancers, treated at a dose of 50 mg/m²/cycle with lifetime cumulative anthracycline doses up to 1,532 mg/m², the incidence of clinically significant cardiac dysfunction was low. Of the 418 patients treated with doxorubicin 50 mg/m²/cycle, and having a baseline measurement of left ventricular ejection fraction (LVEF) and at least one follow-up measurement assessed by MUGA scan, 88 patients had a cumulative anthracycline dose of >400 mg/m², an exposure level associated with an increased risk of cardiovascular toxicity with conventional doxorubicin. Only 13 of these 88 patients (15%) had at least one clinically significant change in their LVEF, defined as an LVEF value less than 45% or a decrease of at least 20 points from baseline. Furthermore, only 1 patient (cumulative anthracycline dose of 944 mg/m²), discontinued study treatment because of clinical symptoms of congestive heart failure.
In a pooled analysis of 4,231 patients receiving doxorubicin for breast cancer, ovarian cancer, multiple myeloma, or AIDS-related KS, ventricular arrythmia, palpitations, cardiac failure, cardiac arrest, bundle branch block right, and ejection fraction decreased were reported uncommonly, and atrioventricular block, cyanosis, and conduction disorder were reported rarely.
As with other DNA-damaging antineoplastic agents, secondary acute myeloid leukemias and myelodysplasias have been reported in patients having received combined treatment with doxorubicin. Therefore, any patient treated with doxorubicin should be kept under haematological supervision.
Although local necrosis following extravasation has been reported very rarely, doxorubicin is considered to be an irritant. Animal studies indicate that administration of doxorubicin hydrochloride as a liposomal formulation reduces the potential for extravasation injury. If any signs or symptoms of extravasation occur (e.g. stinging, erythema) terminate the infusion immediately and restart in another vein. The application of ice over the site of extravasation for approximately 30 minutes may be helpful in alleviating the local reaction. Doxorubicin must not be given by the intramuscular or subcutaneous route.
Recall of skin reaction due to prior radiotherapy has rarely occurred with doxorubicin administration.
Post-marketing experience
Adverse drug reactions identified during the post-marketing experience with doxorubicin are described in Table 5. The frequencies are provided according to the following convention: Very common ≥1/10, Common ≥1/100 and <1/10, Uncommon ≥1/1,000 and <1/100, Rare ≥1/10,000, <1/1,000, Very rare <1/10,000 including isolated reports.
Table 5. Adverse drug reactions identified during the post-marketing experience with doxorubicin:
Neoplasms benign, malignant and unspecified (incl cysts and polyps)
very rare: secondary oral neoplasms1
Vascular disorders
uncommon: venous thromboembolism, including thrombophlebitis, venous thrombosis and pulmonary embolism
Skin and subcutaneous tissue disorders
rare: lichenoid keratosis
very rare: erythema multiforme, Stevens Johnson syndrome and toxic epidermal necrolysis
1 Cases of secondary oral cancer have been reported in patients with long-term (more than one year) exposure to doxorubicin or those receiving a cumulative doxorubicin dose greater than 720 mg/m².
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.