Imidafenacin
Chemical formula: C₂₀H₂₁N₃O Molecular mass: 319.408 g/mol PubChem compound: 6433090
Mechanism of action
Contraction of the urinary bladder is known to be induced by acetylcholine with mediation of muscarinic acetylcholine receptor subtype M3. Acetylcholine release from the nerve terminal of the urinary bladder is probably enhanced by a stimulus to muscarinic acetylcholine receptor subtype M1.
Imidafenacin antagonizes subtypes M3 and M1 in vitro. In the urinary bladder, imidafenacin inhibits acetylcholine release by antagonizing subtype M1 and contraction of smooth muscles by antagonizing subtype M3. Compared with the inhibitory effect on the salivary gland, imidafenacin shows higher inhibitory effect on the urinary bladder contraction, probably indicating efficacy and safety of these products in the clinical practice.
Pharmacodynamic properties
Pharmacological Activity
1) Activity in the muscarinic acetylcholine receptor subtypes (in vitro)
(1) Antagonistic activity of imidafenacin was investigated on muscarinic acetylcholine receptors in vas deferens (M1), atrium (M2), and ileum (M3) using tissue specimens prepared from rabbits and guinea pigs. Imidafenacin showed higher antagonistic activity in the ileum (M3) and vas deferens (M1), compared with atrium (M2). Major metabolites in humans showed no antagonistic activity in the muscarinic acetylcholine receptor subtypes.
(2) Antagonistic activity of imidafenacin was investigated in recombinant human muscarinic acetylcholine receptor subtypes M1, M2, and M3 in the receptor binding assay. Imidafenacin showed high affinities for subtypes M3 and M1.
(3) Imidafenacin inhibited acetylcholine release and urinary bladder contraction by antagonizing subtypes M3 and M1 in the tissue specimens prepared from rats.
2) Activity in the urinary bladder (in vivo)
(1) Imidafenacin decreased rhythmic contraction of the rat urinary bladder dose-dependently.
(2) Imidafenacin inhibited a carbachol-induced decrease in the capacity of the rat urinary bladder dosedependently.
3) Selectivity for the urinary bladder
(1) In rats, the activity ratio of inhibition of rhythmic contraction in the urinary bladder to carbacholinduced salivary secretion was about 10 times higher in imidafenacin than in propiverine hydrochloride, demonstrating high selectivity of imidafenacin for the urinary bladder.
(2) Evaluation of rat performance in the Morris water maze task indicated that antagonistic activity of imidafenacin on subtype M1 was unlikely to impair spatial learning and memory.
Pharmacokinetic properties
Plasma Concentrations
(1) Single administration
1) Effect of meal
After single oral administration of 0.1 mg of imidafenacin to healthy adult males (n=12) at the fasting state, plasma concentration reached the peak (Cmax: 471 pg/mL) at 1.5 hours, and decreased with a half-life of 2.9 hours. Cmax and AUC0-12 at the fed state were about 1.3 and 1.2 times higher than those at the fasting state, respectively.
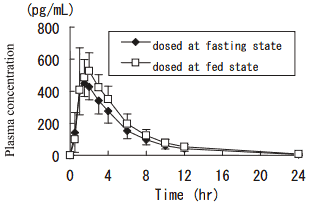
Pharmacokinetic parameters
| Dosing state | Tmax (hr) | Cmax (pg/mL) | AUC0-12 (pg·hr/mL) | T1/2 (hr) |
|---|---|---|---|---|
| Fasting | 1.5 | 471 ± 107 | 2230 ± 540 | 2.9 ± 0.2 |
| Fed | 1.3 | 611 ± 113 | 2690 ± 470 | 2.9 ± 0.2 |
Mean ± S.D. for Cmax, AUC0-12, and T1/2; median for Tmax
2) Bioequivalence study
Biological equivalence of imidafenacin OD Tablets 0.1 mg without water (n=24) or with water (n=24) and imidafenacin Tablets 0.1 mg with water was demonstrated in a cross-over bioequivalence study after single oral administration of these two formulations to healthy adult males at the fasting state.
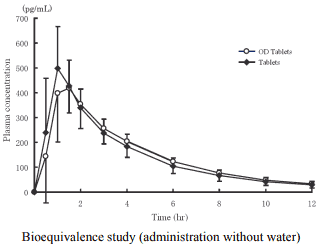
Pharmacokinetic parameters
| Formulation | Tmax (hr) | Cmax (pg/mL) | AUC0-12 (pg·hr/mL) | T1/2 (hr) |
|---|---|---|---|---|
| OD Tablets | 1.4 ± 0.7 | 487 ± 137 | 1830 ± 492 | 3.09 ± 0.46 |
|Tablets|<>1.1 ± 0.3|<>552 ± 140|<>1810 ± 467|<>3.04 ± 0.41
Mean ± S.D
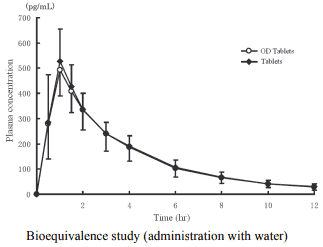
Pharmacokinetic parameters
| Formulation | Tmax (hr) | Cmax (pg/mL) | AUC0-12 (pg·hr/mL) | T1/2 (hr) |
|---|---|---|---|---|
| OD Tablets | 1.0 ± 0.2 | 495 ± 99.8 | 1810 ± 449 | 3.08 ± 0.44 |
| Tablets | 1.0 ± 0.2 | 541 ± 119 | 1860 ± 381 | 3.15 ± 0.52 |
Mean ± S.D.
(2) Repeated administration
After repeated oral administration of 0.25 mg of imidafenacin twice daily for 5 days to the healthy adult males (n=5), the time-course of plasma concentration and pharmacokinetic parameters after the final dosing were comparable to those after the initial dosing, indicating no accumulation of imidafenacin after repeated administration.
Note) The dosage in this study is different from the approved dosage and administration of these products.
(3) The elderly
After single oral administration of 0.1 mg of imidafenacin to the healthy non-elderly adult males (n=6) and the elderly males aged 65 years or more (n=9) at the fasting state, Cmax in the elderly was about 1.2 times higher than that in the non-elderly, while AUC0-∞ was comparable between the two groups.
| Tmax (hr) | Cmax (pg/mL) | AUC0-∞ (pg·hr/mL) | T1/2 (hr) | |
|---|---|---|---|---|
| Non-elderly | 1.5 | 382 ± 106 | 2010 ± 1050 | 2.6 ± 0.7 |
| Elderly | 1.0 | 445 ± 136 | 2140 ± 480 | 3.1 ± 0.4 |
Mean ± S.D. for Cmax, AUC0-∞, and T1/2; median for Tmax
(4) Population pharmacokinetic (PPK) analysis
A two-compartment model involving primary absorption with a lag time in absorption was used for the analysis of population pharmacokinetics by NONMEM. Plasma concentration of imidafenacin was determined at a total of 3168 points in 852 patients with overactive bladder aged 20 to 85 years (including 101 patients with mild hepatic dysfunction, 116 patients with mild renal dysfunction, and 14 patients with moderate renal dysfunction) and 90 healthy adults aged 20 to 75 years in the long-term study and long-term ascending-dose study. The relationship of clearance (CL/F) of imidafenacim to the following covariates was assessed: body weight, age, gender, drinking habit, smoking habit, indices for hepatic function (AST [GOT], ALT [GPT], γ-GTP, ALP, lactate dehydrogenase, and total bilirubin), indices for renal function (serum creatinine, and blood urea nitrogen), and albumin. CL/F in the patients with mild abnormality in ALP was lower than that in the normal patients by 4%. CL/F in the elderly was lower than that in the non-elderly by 14%. The other covariates including indices for renal function (serum creatinine, blood urea nitrogen) did not affect CL/F.
| Population parameter | Estimate (95% CI) | Interindividual variability |
|---|---|---|
| Total body clearance (L/hr) | 23.1 (21.2 to 25.0) | 32.4% |
| Volume of distribution for the central compartment (L) | 109 (102 to 116) | 23.3% |
| Inter-compartmental clearance (L/hr) | 3.50 (2.95 to 4.05) | |
| Volume of distribution for the peripheral compartment (L) | 44.3 (33.8 to 54.8) | |
| Rate constant for absorption (1/hr) | 3.07 (2.55 to 3.59) | 136.7% |
| Absorption lag time (hr) | 0.436 (0.422 to 0.450) | |
| Intra-individual variability | 37.3% |
There is no clinical experience to administer 0.2 and 0.4 mg/day of imidafenacin to patients with moderate to severe hepatic dysfunction and those with severe renal dysfunction in the clinical studies including the long-term ascending-dose study.
Absorption (for reference: overseas data)
In the healthy adult foreign males, imidafenacin was absorbed almost 100% from the gastrointestinal tract, with an absolute bioavailability of 57.8%.
Metabolism
After oral administration, about 40% of imidafenecin was subjected to first-pass effect in the liver. Major plasma metabolites included M-2 (oxidized metabolite on the imidazole ring of imidafenacin), M-4 (ring-cleaved metabolite of M-2), and M-9 (N-glucuronide of imidafenacin). Metabolism to M-2 and M-4 was primarily catalyzed by CYP3A4, and that to M-9 was by UGT1A4.
In addition, imidafenacin and its major metabolites, M-2, M-4, and M-9, did not inhibit human CYP species in vitro (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4).
Excretion (for reference: overseas data)
After single oral administration of 14C-imidafenacin to healthy adult foreign males (n=6) at a dose of 0.25 mg at the fasting state, 95% of the dose was recovered as radioactivity in the urine and feces until 192 hours after administration (65.6% in the urine, and 29.4% in the feces). Less than 10% of the dose was excreted unchanged in the urine, and none of the dose was excreted unchanged in the feces.
Note) The dosage in this study is different from the approved dosage and administration of these products.
Protein Binding
The protein binding ratio of imidafenacin ranged from 87.1 to 88.8%. Major binding proteins were albumin and α1-acid glycoprotein.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.